0001832038false00018320382024-10-292024-10-29
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 8-K
CURRENT REPORT
Pursuant to Section 13 or 15(d)
of the Securities Exchange Act of 1934
Date of report (Date of earliest event reported): October 29, 2024
Invivyd, Inc.
(Exact Name of Registrant as Specified in its Charter)
|
|
|
|
|
|
|
|
|
|
Delaware |
|
001-40703 |
|
85-1403134 |
(State or Other Jurisdiction of Incorporation) |
|
(Commission File Number) |
|
(IRS Employer Identification No.) |
|
|
|
1601 Trapelo Road, Suite 178 Waltham, MA |
|
02451 |
(Address of Principal Executive Offices) |
|
(Zip Code) |
Registrant’s telephone number, including area code: (781) 819-0080
Not applicable
(Former Name or Former Address, if Changed Since Last Report)
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions:
|
|
☐ |
Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425) |
|
|
☐ |
Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12) |
|
|
☐ |
Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b)) |
|
|
☐ |
Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c)) |
Securities registered pursuant to Section 12(b) of the Act:
|
|
|
|
|
Title of each class |
|
Trading Symbol(s) |
|
Name of each exchange on which registered |
Common stock, par value $0.0001 per share |
|
IVVD |
|
The Nasdaq Stock Market LLC |
Indicate by check mark whether the registrant is an emerging growth company as defined in Rule 405 of the Securities Act of 1933 (§230.405 of this chapter) or Rule 12b-2 of the Securities Exchange Act of 1934 (§240.12b-2 of this chapter).
Emerging growth company ☒
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
|
|
Item 2.02. |
Results of Operations and Financial Condition. |
On October 29, 2024, Invivyd, Inc. (the “Company”) issued a press release entitled “Invivyd Reports Preliminary Third Quarter 2024 Results, Withdraws Prior Financial Guidance, and Targets Near-Term Profitability” (the “Press Release”), which included the Company’s estimated third quarter 2024 PEMGARDA™ (pemivibart) net product revenue and estimated cash and cash equivalents as of September 30, 2024. The amounts included in the Press Release are preliminary and are subject to change upon completion of the Company’s financial closing procedures for the quarter ended September 30, 2024, and finalization of the unaudited interim condensed consolidated financial statements. The preliminary financial data included in the Press Release has been prepared by, and is the responsibility of, the Company’s management. PricewaterhouseCoopers LLP, the Company’s independent registered public accounting firm, has not audited, reviewed, examined, compiled, nor applied agreed-upon procedures with respect to the preliminary financial data. Accordingly, PricewaterhouseCoopers LLP does not express an opinion or any other form of assurance with respect thereto. Additional information and disclosures would be required for a more complete understanding of the Company’s financial position and results of operations as of September 30, 2024. A copy of the Press Release is filed herewith as Exhibit 99.1 and is incorporated by reference in this Item 2.02.
On October 29, 2024, the Company issued the Press Release, a copy of which is filed herewith as Exhibit 99.1 and is incorporated by reference in this Item 8.01.
On October 29, 2024, the Company issued a press release entitled “Invivyd Phase 3 Long-Term Exploratory Clinical Efficacy Data Shows PEMGARDA™ (pemivibart) Provided Substantial Protection From Symptomatic COVID-19 Versus Placebo Over Six Months of Follow-Up, With No Additional Doses, In Immunocompetent Participants.” A copy of the press release is filed herewith as Exhibit 99.2 and is incorporated by reference in this Item 8.01.
|
|
Item 9.01. |
Financial Statements and Exhibits. |
(d) Exhibits
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, the registrant has duly caused this report to be signed on its behalf by the undersigned hereunto duly authorized.
INVIVYD, INC.
Date: October 29, 2024 By: /s/ Jill Andersen
Jill Andersen
Chief Legal Officer and Corporate Secretary

Invivyd Reports Preliminary Third Quarter 2024 Results, Withdraws Prior Financial Guidance, and Targets Near-Term Profitability
•Preliminary Q3 2024 PEMGARDA™ (pemivibart) net product revenue of $9.3 million; Invivyd ended Q3 2024 with approximately $107 million in cash and cash equivalents
•Expects to finish 2024 with $65 million or more in cash and cash equivalents
•Withdraws formal revenue guidance due to recent growth headwind from U.S. FDA’s late-Q3 2024 warning on potential for substantially reduced activity of pemivibart through the PEMGARDA Fact Sheet and other media based on contested, third-party, non-peer-reviewed, non-reproducible, virologic activity data from a non-pemivibart antibody
•Targets near-term (1H 2025) profitability with existing cash and cash equivalents, anticipated growth of net product revenue, and various operational efficiency improvements
•Further update to be provided on Q3 2024 earnings call
WALTHAM, Mass., Oct. 29, 2024 -- Invivyd, Inc. (Nasdaq: IVVD), a biopharmaceutical company devoted to delivering protection from serious viral infectious diseases, today reported preliminary third quarter (Q3) 2024 financial results and an update to prior financial guidance.
Invivyd previously guided to $150-200 million in PEMGARDA™ (pemivibart) net product revenue in 2024 and expected to finish the year with $75 million or more in cash and cash equivalents. This previous guidance did not contemplate the impact on product growth at the most critical moment in Invivyd’s financial year arising from the U.S. Food and Drug Administration (FDA) updating the PEMGARDA Fact Sheet for Healthcare Providers (HCPs) (Fact Sheet) in late-Q3 2024 to include a link to contested, third-party, non-peer-reviewed, non-reproducible, virologic activity data from a non-pemivibart antibody and convey that, based on such data, PEMGARDA may have substantially reduced susceptibility to the currently circulating SARS-CoV-2 variants (i.e., KP.3.1.1). Invivyd previously communicated this unfortunate and unnecessary action that created doubt regarding the antiviral activity of PEMGARDA against current circulating variants. This resulted in confusion in the HCP and vulnerable population communities and an uncertainty regarding the pursuit of additional options for protection against COVID-19 for certain immunocompromised people.
Though not at previously anticipated rates, PEMGARDA net product revenue grew through the third quarter and continues to grow. Now with a corrected PEMGARDA Fact Sheet
removing the aforementioned data reference and including authentic pemivibart data reflecting ongoing neutralization activity against currently circulating variants tested in line with prior variants represented in the CANOPY Phase 3 clinical trial, Invivyd expects ongoing commercial optimization to drive growth.
Based on currently available information, the company is announcing preliminary Q3 2024 PEMGARDA net product revenue of $9.3 million. The company estimates that Invivyd ended Q3 2024 with approximately $107 million in cash and equivalents, and, with anticipated growth of net product revenue and various operational efficiencies underway, believes existing cash and cash equivalents will be sufficient to fund operations through profitability. These estimates are preliminary and are subject to change upon completion of the company’s financial closing procedures for Q3 2024, and finalization of the unaudited interim condensed consolidated financial statements.
“At the Infectious Disease (ID) Week meeting in mid-October, we were thrilled to engage with healthcare professionals in an effort to alleviate any remaining confusion regarding the antiviral activity of PEMGARDA, and to share the positive, CANOPY clinical trial data and the safety data included in the Fact Sheet,” said Tim Lee, Chief Commercial Officer of Invivyd. “Today’s announcement of a new exploratory analysis from the CANOPY Phase 3 clinical trial showing that PEMGARDA provided substantial protection from symptomatic COVID-19 versus placebo in immunocompetent participants through month twelve, absent additional dosing and with minimal drug concentration, only amplifies our confidence about re-accelerating our product growth in COVID-19 pre-exposure prophylaxis (PreP) in the appropriate immunocompromised population. It also reflects our potential to deliver meaningful protection to certain immunocompromised adults and adolescents who may not mount a strong enough immune response to vaccines alone. This population must not be forgotten.”
“We continue to feel confident about the demand for PEMGARDA and our ability to return to strong product growth despite the unnecessary headwinds inflicted by the Agency,” said Marc Elia, Chairman of the Invivyd Board of Directors. “While we are engaged in discussions with key leaders at the FDA, it is clear that we must also focus on what matters most: the vulnerable populations who deserve more protection against this devastating virus, especially as we enter the winter season where many individuals hope to gather with friends and family. Our confidence in the ultimate commercial potential of PEMGARDA is unchanged.”
About Invivyd
Invivyd, Inc. (Nasdaq: IVVD) is a biopharmaceutical company devoted to delivering protection from serious viral infectious diseases, beginning with SARS-CoV-2. The company’s proprietary INVYMAB™ platform approach combines state-of-the-art viral
surveillance and predictive modeling with advanced antibody engineering. INVYMAB is designed to facilitate the rapid, serial generation of new monoclonal antibodies (mAbs) to address evolving viral threats. In March 2024, Invivyd received emergency use authorization (EUA) from the U.S. FDA for its first mAb in a planned series of innovative antibody candidates. Visit https://invivyd.com/ to learn more.
About PEMGARDA
PEMGARDA™ (pemivibart) is a half-life extended investigational monoclonal antibody (mAb). PEMGARDA was engineered from adintrevimab, Invivyd’s investigational mAb that has a robust safety data package and provided evidence of clinical efficacy in a global Phase 2/3 clinical trial for the prevention and treatment of COVID-19. PEMGARDA has demonstrated in vitro neutralizing activity against major SARS-CoV-2 variants, including JN.1. PEMGARDA targets the SARS-CoV-2 spike protein receptor binding domain (RBD), thereby inhibiting virus attachment to the human ACE2 receptor on host cells.
PEMGARDA (pemivibart) injection (4500 mg), for intravenous use is an investigational mAb that has not been approved, but has been authorized for emergency use by the U.S. FDA under an EUA for the pre-exposure prophylaxis (prevention) of COVID-19 in adults and adolescents (12 years of age and older weighing at least 40 kg) who have moderate-to-severe immune compromise due to certain medical conditions or receipt of certain immunosuppressive medications or treatments and are unlikely to mount an adequate immune response to COVID-19 vaccination. Recipients should not be currently infected with or have had a known recent exposure to an individual infected with SARS-CoV-2.
PEMGARDA is not authorized for use for treatment of COVID-19 or post-exposure prophylaxis of COVID-19. Pre-exposure prophylaxis with PEMGARDA is not a substitute for vaccination in individuals for whom COVID-19 vaccination is recommended. Individuals for whom COVID-19 vaccination is recommended, including individuals with moderate-to-severe immune compromise who may derive benefit from COVID-19 vaccinations, should receive COVID-19 vaccination. In individuals who have recently received a COVID-19 vaccine, PEMGARDA should be administered at least 2 weeks after vaccination.
Anaphylaxis has been observed with PEMGARDA and the PEMGARDA Fact Sheet for Healthcare Providers includes a boxed warning for anaphylaxis. The most common adverse events (all grades, incidence ≥2%) observed in participants who have moderate-to-severe immune compromise treated with PEMGARDA included systemic and local infusion-related or hypersensitivity reactions, upper respiratory tract infection, viral infection, influenza-like illness, fatigue, headache, and nausea. For additional information, please see the PEMGARDA full product Fact Sheet for Healthcare Providers, including important safety information and boxed warning.
To support the EUA for PEMGARDA, an immunobridging approach was used to determine if PEMGARDA may be effective for pre-exposure prophylaxis of COVID-19. Immunobridging is based on the serum virus neutralizing titer-efficacy relationships identified with other
neutralizing human mAbs against SARS-CoV-2. This includes adintrevimab, the parent mAb of pemivibart, and other mAbs that were previously authorized for EUA. There are limitations of the data supporting the benefits of PEMGARDA. Evidence of clinical efficacy for other neutralizing human mAbs against SARS-CoV-2 was based on different populations and SARS-CoV-2 variants that are no longer circulating. Further, the variability associated with cell-based EC50 value determinations, along with limitations related to pharmacokinetic data and efficacy estimates for the mAbs in prior clinical trials, impact the ability to precisely estimate protective titer ranges. Additionally, certain SARS-CoV-2 viral variants may emerge that have substantially reduced susceptibility to PEMGARDA, and PEMGARDA may not be effective at preventing COVID-19 caused by these SARS-CoV-2 viral variants.
The emergency use of PEMGARDA is only authorized for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of drugs and biological products during the COVID-19 pandemic under Section 564(b)(1) of the Federal Food, Drug, and Cosmetic Act, 21 U.S.C. § 360bbb-3(b)(1), unless the declaration is terminated or authorization revoked sooner. PEMGARDA is authorized for use only when the combined national frequency of variants with substantially reduced susceptibility to PEMGARDA is less than or equal to 90%, based on available information including variant susceptibility to PEMGARDA and national variant frequencies.
Cautionary Note Regarding Forward Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as “anticipates,” “believes,” “could,” “expects,” “estimates,” “intends,” “potential,” “preliminary,” “projects,” and “future” or similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. Forward-looking statements include statements concerning, among other things, the company’s preliminary third quarter 2024 results; the company’s expectation regarding its cash and cash equivalents balance at the end of 2024; the company’s aim for near-term profitability, expectations regarding the commercialization of PEMGARDA, and plans for operational efficiency improvements; the company’s belief that its existing cash and cash equivalents, and anticipated growth of net product revenue and various operational efficiencies, will be sufficient to fund operations through profitability; the company’s expectations regarding its interactions with the FDA; the company’s ongoing research and clinical development activities, as well as future potential research and clinical development efforts; the company’s expectations regarding potential PEMGARDA product growth in the appropriate immunocompromised population, and the company’s potential to deliver meaningful protection to certain immunocompromised adults and adolescents who may not mount a strong enough immune response to vaccines alone; the company’s belief in the ultimate commercial potential of PEMGARDA; the potential of PEMGARDA as a mAb for pre-exposure prophylaxis (prevention) of COVID-19 in certain adults and adolescents who have moderate-to-severe immune compromise; the company’s devotion
to delivering protection from serious viral infectious diseases, beginning with SARS-CoV-2; the design of the company’s INVYMAB platform approach to facilitate the rapid, serial generation of new mAbs to address evolving viral threats; the company’s business strategies and objectives; and other statements that are not historical fact. The company may not actually achieve the plans, intentions or expectations disclosed in the company’s forward-looking statements and you should not place undue reliance on the company’s forward-looking statements. These forward-looking statements involve risks and uncertainties that could cause the company’s actual results to differ materially from the results described in or implied by the forward-looking statements, including, without limitation: uncertainties regarding the company’s expectations, projections and estimates regarding future costs and expenses, future revenue, capital requirements, and the availability of and the need for additional financing; whether the company’s cash and cash equivalents are sufficient to support its operating plan for as long as anticipated; uncertainties regarding market acceptance, payor coverage or future sales and revenue generated by PEMGARDA; uncertainties regarding the potential advantages from the company’s planned operational efficiency improvements; the timing, progress and results of the company’s discovery, preclinical and clinical development activities; the risk that results of nonclinical studies or clinical trials may not be predictive of future results, and interim data are subject to further analysis; unexpected safety or efficacy data observed during preclinical studies or clinical trials; the predictability of clinical success of the company’s product candidates based on neutralizing activity in nonclinical studies; potential variability in neutralizing activity of product candidates tested in different assays, such as pseudovirus assays and authentic assays; the company’s reliance on third parties with respect to virus assay creation and product candidate testing and with respect to its clinical trials; variability of results in models used to predict activity against SARS-CoV-2 variants; whether pemivibart or any other product candidate is able to demonstrate and sustain neutralizing activity against major SARS-CoV-2 variants, particularly in the face of viral evolution; how long the EUA granted by the FDA for PEMGARDA will remain in effect and whether the EUA is revised or revoked by the FDA; the company’s ability to build and maintain sales, marketing and distribution capabilities to successfully commercialize PEMGARDA; uncertainties related to the regulatory authorization or approval process, and available development and regulatory pathways for authorization or approval of the company’s product candidates; the ability to maintain a continued acceptable safety, tolerability and efficacy profile of any product candidate following regulatory authorization or approval; changes in the regulatory environment; changes in expected or existing competition; the complexities of manufacturing mAb therapies; the company’s ability to leverage its INVYMAB platform approach to facilitate the rapid, serial generation of new mAbs to address evolving viral threats; any legal proceedings or investigations relating to the company; any change in the preliminary estimates of the company’s third quarter 2024 results upon completion of the company’s financial closing procedures, and finalization of the unaudited interim condensed consolidated financial statements; the company’s ability to continue as a going concern; and whether the company has adequate funding to meet future operating expenses and capital expenditure requirements. Other factors that may cause the company’s actual results to differ materially from those expressed or implied in
the forward-looking statements in this press release are described under the heading “Risk Factors” in the company’s Annual Report on Form 10-K for the year ended December 31, 2023 and the company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2024, each filed with the Securities and Exchange Commission (SEC), and in the company’s other filings with the SEC, and in its future reports to be filed with the SEC and available at . Forward-looking statements contained in this press release are made as of this date, and Invivyd undertakes no duty to update such information whether as a result of new information, future events or otherwise, except as required under applicable law.
This press release contains hyperlinks to information that is not deemed to be incorporated by reference in this press release.
Contacts:
Media Relations
(781) 208-1747
media@invivyd.com
Investor Relations
(781) 208-1747
investors@invivyd.com

Invivyd Phase 3 Long-Term Exploratory Clinical Efficacy Data Shows PEMGARDA™ (pemivibart) Provided Substantial Protection from Symptomatic COVID-19 Versus Placebo Over Six Months of Follow-Up, With No Additional Doses, In Immunocompetent Participants
•Following strong protection (84% relative risk reduction versus placebo) demonstrated through month 6 with pemivibart, CANOPY clinical trial participants were followed for an additional six-month period (months 7-12), with no additional doses, to assess safety and efficacy as drug concentrations declined over time
•In the six-month off-drug follow-up period (months 7-12), substantially reduced concentrations of PEMGARDA reduced the risk of symptomatic COVID-19 by 64% compared to placebo in an immunocompetent adult population, for total twelve-month risk reduction of 76% following two initial pemivibart doses (nominal p <0.0001)
•Months 7-12 follow-up period included the U.S. summer KP.3 and KP.3.1.1 dominant wave, per CDC surveillance
•No new safety observations occurred months 7 - 12
•Across all 12 months of the CANOPY clinical trial, Cohort B placebo arm participants experienced an 18% rate of PCR-confirmed symptomatic COVID-19, excluding reinfections, indicative of continued, widespread, uncontrolled virus transmission in the U.S.
WALTHAM, Mass., Oct. 29, 2024 -- Invivyd, Inc. (Nasdaq: IVVD), a biopharmaceutical company devoted to delivering protection from serious viral infectious diseases, today announced positive 12-month exploratory clinical efficacy data from the company’s ongoing CANOPY Phase 3 clinical trial of pemivibart, a half-life extended investigational monoclonal antibody (mAb), for the pre-exposure prophylaxis (PrEP) of COVID-19.
The exploratory clinical efficacy data from follow-up months 7-12 in CANOPY Cohort B, a placebo-controlled cohort of immunocompetent individuals at risk for symptomatic COVID-19 disease due to regular unmasked face-to-face indoor interactions, showed a relative risk reduction (RRR) of 64% in the pemivibart arm compared to placebo in the likelihood of trial participants contracting confirmed symptomatic COVID-19. Continued protection over this follow-up period, absent continued dosing, was a prespecified exploratory endpoint and, added to initial 180-day clinical efficacy observations, generated an overall 12-month protection rate of 76% following two doses of pemivibart (administered on Days 1 and 90), nominally significant at <0.0001. Clinical efficacy data are displayed in the table below.
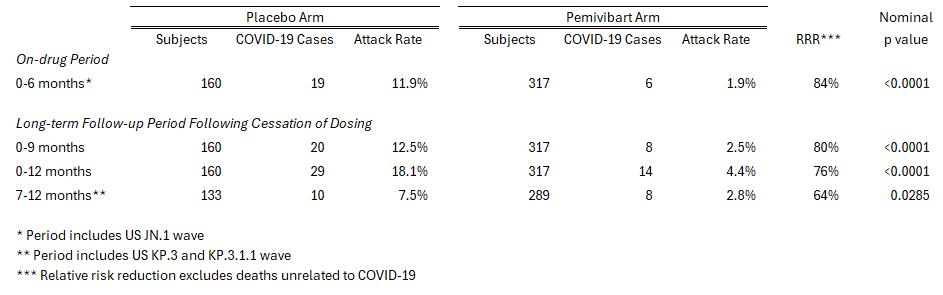
Pemivibart has an estimated in vivo half-life of approximately 45 days, and pemivibart IC50 values against circulating viruses are tested routinely, allowing for the calculation of the average serum virus neutralizing antibody (sVNA) titer over time, the measure of how much antiviral activity pemivibart confers to treated participants. Previously published literature1 indicates likely robust protection from symptomatic COVID-19 at sVNA titer levels well below the levels delivered by the authorized PEMGARDA (pemivibart) dosing regimen. In this updated CANOPY exploratory analysis, the 64% RRR (excluding deaths unrelated to COVID-19) that residual circulating pemivibart delivered versus placebo in months 7 through 12 occurred at calculated average titers of only approximately 7% of the average titers during the active dosing period. This was attributable to the expected decline in pemivibart serum concentrations following cessation of dosing, along with modest change to assessed pemivibart IC50 in the transition from the JN.1 dominant virus to the KP lineage dominant circulating viruses.
“Today’s data underline our high confidence in the clinical benefit that mAbs like pemivibart can confer even at very low serum concentrations and associated antiviral titers,” commented Robert Allen, Ph.D., Chief Scientific Officer of Invivyd. “Engineered mAbs are designed to confer a step change in protection compared to the protection possible from native immune response to infection and vaccination boosts2. We believe that the clinical risk reduction generated at such low titers in this CANOPY clinical trial follow-up, which builds on now longstanding published data, implies that the modest changes (less than ~10 fold change) in pemivibart IC50 observed against emerging variants are of questionable significance.”
“Our company’s goal is to provide high quality options for protection from SARS-CoV-2. Today’s exploratory clinical efficacy data update from our Phase 3 trial is critical, as it adds contemporary context to longstanding data showing strong protection from COVID-19
1Schmidt, et al. Antibody-Mediated Protection Against Symptomatic COVID-19 Can Be Achieved at Low Serum Neutralizing Titers. Sci. Transl. Med. 15, eadg2783 (2023); Stadler, et al. Monoclonal Antibody Levels and Protection From COVID-19, Nat. Commun., 14:4545 (2023)
2 https://www.cdc.gov/acip/downloads/slides-2024-06-26-28/03-COVID-Link-Gelles-508.pdf
disease at relatively low sVNA titers with mAbs,” commented Marc Elia, Chairman of the Invivyd Board of Directors. “These consistent observations now span years and countless virus lineages and mutations, and, in toto, suggest that Invivyd, pending alignment with regulators, can develop and commercialize low-dose, high potency, long-acting mAbs that can have the same route of administration and system-friendliness as COVID-19 vaccine boosts, but with the goal of higher, more consistent, and longer-acting protection. Our current pipeline molecule, VYD2311, is currently in Phase 1 testing including an intramuscular (IM) administration in anticipation of executing against this exact goal.”
The safety profile of pemivibart over the 12-month study period was reassuring with no new trends or safety signals observed for treatment-emergent adverse events (TEAEs) or infusion-related reactions or hypersensitivity reactions since the disclosure of the 6-month CANOPY data. Over the 12 months, in Cohort A, an open-label single-arm cohort, the most common TEAEs (>5%) included upper respiratory tract infection (URTI) (10.1%), viral infection (8.8%), urinary tract infection (UTI) (5.6%), and influenza-like illness (5.2%). As previously disclosed, anaphylaxis was observed in 4 participants (0.6%) in Cohort A – 2 participants during the first infusion and 2 participants during the second infusion; two reactions were life-threatening, and all led to permanent discontinuation of pemivibart. Infusion-related reactions and hypersensitivity reactions within 24 hours of dosing pemivibart were observed in 8.2% and 3.9% of participants in Cohort A after the initial dose and redose, respectively, and were generally mild to moderate in severity. Over the 12 months, in Cohort B, the most common TEAEs in the pemivibart arm included viral infection (8.5%), URTI (8.5%), and influenza-like illness (5.7%), with higher percentages in the placebo arm. No participants developed anaphylaxis. Infusion-related reactions and hypersensitivity reactions within 24 hours of dosing pemivibart were observed in 1.3% and 2.5% of participants in Cohort B after initial dose and redose, respectively, and were all mild or moderate in severity. Serious Adverse Events (SAEs) occurring in >1 Cohort A participant included pneumonia (n=3), anaphylactic reaction (n=2), hypotension (n=2), pyelonephritis (n=2), syncope (n=2), and UTI (n=2). No SAEs occurred in >1 participant in either treatment arm in Cohort B. These data support that the safety profile for pemivibart remains consistent with the PEMGARDA Fact Sheet for Healthcare Providers and the known safety profile of other SARS-CoV-2-directed mAbs.
CANOPY Cohort B exploratory clinical efficacy results from months 7-12 provide data from reduced concentration of pemivibart and reduced sVNA titers in a contemporary immunocompetent population that had acquired prior immune exposure from either vaccination or natural infection and overlapped with the height of the Summer 2024 KP.3 and KP3.1.1 wave that saw a surge in COVID-19 cases nationwide in the U.S. By contrast, ancestral clinical trials of prior COVID-19 PrEP candidate mAbs were performed in populations naïve to vaccination or infection.
The months’ 7-12 CANOPY Cohort B clinical efficacy exploratory data announced today complement the 180-day clinical efficacy exploratory data demonstrating potential signals
of clinical protection from symptomatic COVID-19 shared previously. The company expects the full data set to be provided in an upcoming scientific publication.
About Invivyd
Invivyd, Inc. (Nasdaq: IVVD) is a biopharmaceutical company devoted to delivering protection from serious viral infectious diseases, beginning with SARS-CoV-2. The company’s proprietary INVYMAB™ platform approach combines state-of-the-art viral surveillance and predictive modeling with advanced antibody engineering. INVYMAB is designed to facilitate the rapid, serial generation of new monoclonal antibodies (mAbs) to address evolving viral threats. In March 2024, Invivyd received emergency use authorization (EUA) from the U.S. FDA for its first mAb in a planned series of innovative antibody candidates. Visit https://invivyd.com/ to learn more.
About PEMGARDA
PEMGARDA™ (pemivibart) is a half-life extended investigational monoclonal antibody (mAb). PEMGARDA was engineered from adintrevimab, Invivyd’s investigational mAb that has a robust safety data package and provided evidence of clinical efficacy in a global Phase 2/3 clinical trial for the prevention and treatment of COVID-19. PEMGARDA has demonstrated in vitro neutralizing activity against major SARS-CoV-2 variants, including JN.1. PEMGARDA targets the SARS-CoV-2 spike protein receptor binding domain (RBD), thereby inhibiting virus attachment to the human ACE2 receptor on host cells.
PEMGARDA (pemivibart) injection (4500 mg), for intravenous use is an investigational mAb that has not been approved, but has been authorized for emergency use by the U.S. FDA under an EUA for the pre-exposure prophylaxis (prevention) of COVID-19 in adults and adolescents (12 years of age and older weighing at least 40 kg) who have moderate-to-severe immune compromise due to certain medical conditions or receipt of certain immunosuppressive medications or treatments and are unlikely to mount an adequate immune response to COVID-19 vaccination. Recipients should not be currently infected with or have had a known recent exposure to an individual infected with SARS-CoV-2.
PEMGARDA is not authorized for use for treatment of COVID-19 or post-exposure prophylaxis of COVID-19. Pre-exposure prophylaxis with PEMGARDA is not a substitute for vaccination in individuals for whom COVID-19 vaccination is recommended. Individuals for whom COVID-19 vaccination is recommended, including individuals with moderate-to-severe immune compromise who may derive benefit from COVID-19 vaccinations, should receive COVID-19 vaccination. In individuals who have recently received a COVID-19 vaccine, PEMGARDA should be administered at least 2 weeks after vaccination.
Anaphylaxis has been observed with PEMGARDA and the PEMGARDA Fact Sheet for Healthcare Providers includes a boxed warning for anaphylaxis. The most common adverse events (all grades, incidence ≥2%) observed in participants who have moderate-to-severe immune compromise treated with PEMGARDA included systemic and local
infusion-related or hypersensitivity reactions, upper respiratory tract infection, viral infection, influenza-like illness, fatigue, headache, and nausea. For additional information, please see the PEMGARDA full product Fact Sheet for Healthcare Providers, including important safety information and boxed warning.
To support the EUA for PEMGARDA, an immunobridging approach was used to determine if PEMGARDA may be effective for pre-exposure prophylaxis of COVID-19. Immunobridging is based on the serum virus neutralizing titer-efficacy relationships identified with other neutralizing human mAbs against SARS-CoV-2. This includes adintrevimab, the parent mAb of pemivibart, and other mAbs that were previously authorized for EUA. There are limitations of the data supporting the benefits of PEMGARDA. Evidence of clinical efficacy for other neutralizing human mAbs against SARS-CoV-2 was based on different populations and SARS-CoV-2 variants that are no longer circulating. Further, the variability associated with cell-based EC50 value determinations, along with limitations related to pharmacokinetic data and efficacy estimates for the mAbs in prior clinical trials, impact the ability to precisely estimate protective titer ranges. Additionally, certain SARS-CoV-2 viral variants may emerge that have substantially reduced susceptibility to PEMGARDA, and PEMGARDA may not be effective at preventing COVID-19 caused by these SARS-CoV-2 viral variants.
The emergency use of PEMGARDA is only authorized for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of drugs and biological products during the COVID-19 pandemic under Section 564(b)(1) of the Federal Food, Drug, and Cosmetic Act, 21 U.S.C. § 360bbb-3(b)(1), unless the declaration is terminated or authorization revoked sooner. PEMGARDA is authorized for use only when the combined national frequency of variants with substantially reduced susceptibility to PEMGARDA is less than or equal to 90%, based on available information including variant susceptibility to PEMGARDA and national variant frequencies.
About CANOPY
The ongoing CANOPY Phase 3 clinical trial is designed to evaluate the safety and tolerability of pemivibart and to assess immunobridging from pemivibart to certain historical data from the company’s previous Phase 2/3 clinical trial of adintrevimab (ADG20) for the prevention of symptomatic COVID-19 (EVADE). Additionally, there are pre-specified exploratory endpoints through three, six and twelve months to evaluate clinical efficacy of pemivibart compared to placebo in the prevention of RT-PCR-confirmed symptomatic COVID-19. The latest analysis from the Phase 3 CANOPY clinical trial includes 365-day data. The CANOPY clinical trial enrolled participants in two cohorts: Cohort A is a single-arm, open-label trial in adults who have moderate-to-severe immune compromise including complex underlying medical conditions. Cohort B is a randomized, placebo-controlled cohort that enrolled adults without moderate-to-severe immune compromise who are at risk of acquiring COVID-19 due to regular unmasked face-to-face interactions in indoor settings.
Cautionary Note Regarding Forward Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as “anticipates,” “believes,” “could,” “expects,” “estimates,” “intends,” “potential,” “projects,” and “future” or similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. Forward-looking statements include statements concerning, among other things, the company’s ongoing research and clinical development activities, as well as future potential research and clinical development efforts; the potential clinical benefit that mAbs like PEMGARDA (pemivibart) can confer, even at very low serum concentrations and associated antiviral titers; the possibility for Invivyd to develop and commercialize low-dose, high potency, long-acting mAbs with the same route of administration and system-friendliness as COVID-19 vaccine boosts, but higher, more consistent, and longer-acting protection; the company’s expectation to provide a full data set from the CANOPY clinical trial in an upcoming scientific publication; the potential of PEMGARDA as a mAb for pre-exposure prophylaxis (prevention) of COVID-19 in certain adults and adolescents who have moderate-to-severe immune compromise; the company’s devotion to delivering protection from serious viral infectious diseases, beginning with SARS-CoV-2; the design of the company’s INVYMAB platform approach to facilitate the rapid, serial generation of new mAbs to address evolving viral threats; the company’s plans for a series of innovative antibody candidates and its goals for pipeline molecule VYD2311; and other statements that are not historical fact. The company may not actually achieve the plans, intentions or expectations disclosed in the company’s forward-looking statements and you should not place undue reliance on the company’s forward-looking statements. These forward-looking statements involve risks and uncertainties that could cause the company’s actual results to differ materially from the results described in or implied by the forward-looking statements, including, without limitation: the timing, progress and results of the company’s discovery, preclinical and clinical development activities; the risk that results of nonclinical studies or clinical trials may not be predictive of future results, and interim data are subject to further analysis; unexpected safety or efficacy data observed during preclinical studies or clinical trials; the predictability of clinical success of the company’s product candidates based on neutralizing activity in nonclinical studies; potential variability in neutralizing activity of product candidates tested in different assays, such as pseudovirus assays and authentic assays; the company’s reliance on third parties with respect to virus assay creation and product candidate testing and with respect to its clinical trials; variability of results in models used to predict activity against SARS-CoV-2 variants; whether pemivibart, VYD2311, or any other product candidate is able to demonstrate and sustain neutralizing activity against major SARS-CoV-2 variants, particularly in the face of viral evolution; how long the EUA granted by the FDA for PEMGARDA will remain in effect and whether the EUA is revised or revoked by the FDA; the company’s ability to build and maintain sales, marketing and distribution capabilities to successfully commercialize PEMGARDA; uncertainties related to the regulatory authorization or approval process, and available development and regulatory pathways for authorization or approval of the company’s product candidates; the ability to maintain a
continued acceptable safety, tolerability and efficacy profile of any product candidate following regulatory authorization or approval; changes in the regulatory environment; changes in expected or existing competition; the complexities of manufacturing mAb therapies; the company’s ability to leverage its INVYMAB platform approach to facilitate the rapid, serial generation of new mAbs to address evolving viral threats; any legal proceedings or investigations relating to the company; the company’s ability to continue as a going concern; and whether the company has adequate funding to meet future operating expenses and capital expenditure requirements. Other factors that may cause the company’s actual results to differ materially from those expressed or implied in the forward-looking statements in this press release are described under the heading “Risk Factors” in the company’s Annual Report on Form 10-K for the year ended December 31, 2023 and the company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2024, each filed with the Securities and Exchange Commission (SEC), and in the company’s other filings with the SEC, and in its future reports to be filed with the SEC and available at www.sec.gov. Forward-looking statements contained in this press release are made as of this date, and Invivyd undertakes no duty to update such information whether as a result of new information, future events or otherwise, except as required under applicable law.
This press release contains hyperlinks to information that is not deemed to be incorporated by reference in this press release.
Contacts:
Media Relations
(781) 208-1747
media@invivyd.com
Investor Relations
(781) 208-1747
investors@invivyd.com
v3.24.3
| X |
- DefinitionBoolean flag that is true when the XBRL content amends previously-filed or accepted submission.
| Name: |
dei_AmendmentFlag |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:booleanItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionFor the EDGAR submission types of Form 8-K: the date of the report, the date of the earliest event reported; for the EDGAR submission types of Form N-1A: the filing date; for all other submission types: the end of the reporting or transition period. The format of the date is YYYY-MM-DD.
| Name: |
dei_DocumentPeriodEndDate |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:dateItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe type of document being provided (such as 10-K, 10-Q, 485BPOS, etc). The document type is limited to the same value as the supporting SEC submission type, or the word 'Other'.
| Name: |
dei_DocumentType |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:submissionTypeItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAddress Line 1 such as Attn, Building Name, Street Name
| Name: |
dei_EntityAddressAddressLine1 |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:normalizedStringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionAddress Line 2 such as Street or Suite number
| Name: |
dei_EntityAddressAddressLine2 |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:normalizedStringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- Definition
+ References
+ Details
| Name: |
dei_EntityAddressCityOrTown |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:normalizedStringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionCode for the postal or zip code
| Name: |
dei_EntityAddressPostalZipCode |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:normalizedStringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionName of the state or province.
| Name: |
dei_EntityAddressStateOrProvince |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:stateOrProvinceItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionA unique 10-digit SEC-issued value to identify entities that have filed disclosures with the SEC. It is commonly abbreviated as CIK. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection b-2
| Name: |
dei_EntityCentralIndexKey |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:centralIndexKeyItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionIndicate if registrant meets the emerging growth company criteria. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection b-2
| Name: |
dei_EntityEmergingGrowthCompany |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:booleanItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionCommission file number. The field allows up to 17 characters. The prefix may contain 1-3 digits, the sequence number may contain 1-8 digits, the optional suffix may contain 1-4 characters, and the fields are separated with a hyphen.
| Name: |
dei_EntityFileNumber |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:fileNumberItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTwo-character EDGAR code representing the state or country of incorporation.
| Name: |
dei_EntityIncorporationStateCountryCode |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:edgarStateCountryItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe exact name of the entity filing the report as specified in its charter, which is required by forms filed with the SEC. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection b-2
| Name: |
dei_EntityRegistrantName |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:normalizedStringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionThe Tax Identification Number (TIN), also known as an Employer Identification Number (EIN), is a unique 9-digit value assigned by the IRS. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection b-2
| Name: |
dei_EntityTaxIdentificationNumber |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:employerIdItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionLocal phone number for entity.
| Name: |
dei_LocalPhoneNumber |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:normalizedStringItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionBoolean flag that is true when the Form 8-K filing is intended to satisfy the filing obligation of the registrant as pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 13e
-Subsection 4c
| Name: |
dei_PreCommencementIssuerTenderOffer |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:booleanItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionBoolean flag that is true when the Form 8-K filing is intended to satisfy the filing obligation of the registrant as pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 14d
-Subsection 2b
| Name: |
dei_PreCommencementTenderOffer |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:booleanItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTitle of a 12(b) registered security. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection b
| Name: |
dei_Security12bTitle |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:securityTitleItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionName of the Exchange on which a security is registered. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 12
-Subsection d1-1
| Name: |
dei_SecurityExchangeName |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:edgarExchangeCodeItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionBoolean flag that is true when the Form 8-K filing is intended to satisfy the filing obligation of the registrant as soliciting material pursuant to Rule 14a-12 under the Exchange Act. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Exchange Act
-Number 240
-Section 14a
-Subsection 12
| Name: |
dei_SolicitingMaterial |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:booleanItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionTrading symbol of an instrument as listed on an exchange.
| Name: |
dei_TradingSymbol |
| Namespace Prefix: |
dei_ |
| Data Type: |
dei:tradingSymbolItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
| X |
- DefinitionBoolean flag that is true when the Form 8-K filing is intended to satisfy the filing obligation of the registrant as written communications pursuant to Rule 425 under the Securities Act. Reference 1: http://www.xbrl.org/2003/role/presentationRef
-Publisher SEC
-Name Securities Act
-Number 230
-Section 425
| Name: |
dei_WrittenCommunications |
| Namespace Prefix: |
dei_ |
| Data Type: |
xbrli:booleanItemType |
| Balance Type: |
na |
| Period Type: |
duration |
|
Invivyd (NASDAQ:IVVD)
Historical Stock Chart
From Nov 2024 to Dec 2024

Invivyd (NASDAQ:IVVD)
Historical Stock Chart
From Dec 2023 to Dec 2024
